Introduction
Giant cell arteritis (GCA) is the most common primary vasculitis in adults over the age of 50 years.1 Classical symptoms of headache, scalp tenderness and jaw claudication occur when the temporal arteries are affected. Visual loss may occur secondary to arteritic anterior ischaemic optic neuropathy. Other medium-to-large calibre extracranial vessels can be involved, particularly the aorta and its principal branches. Stroke, most commonly in the vertebrobasilar territory, is a rare complication (2–4%) that occurs more commonly in males.2 Multiple imaging modalities maybe useful in the diagnosis of GCA (colour Doppler ultrasound, CT, MRI, PET) but the gold standard remains a temporal artery biopsy demonstrating granulomatous infiltrate in the inner half of the tunica media, particularly the internal elastic membrane. The prompt treatment of suspected GCA with high-dose steroids and aspirin is key to the prevention of ischaemic complications. This case report describes an unusual presentation of GCA with bilateral cerebellar peduncle infarcts refractory to empirical treatment.
Case presentation
A 72-year-old male presented with a 10-month history of scalp tenderness, jaw claudication and general malaise. There were no systemic features such as night sweats, change in appetite or weight. Past medical history included hypertension, hiatus hernia, and a history of high alcohol intake and smoking. Detailed systemic and neurological examination including ophthalmological assessment were normal. Both temporal arteries were palpable and nontender.
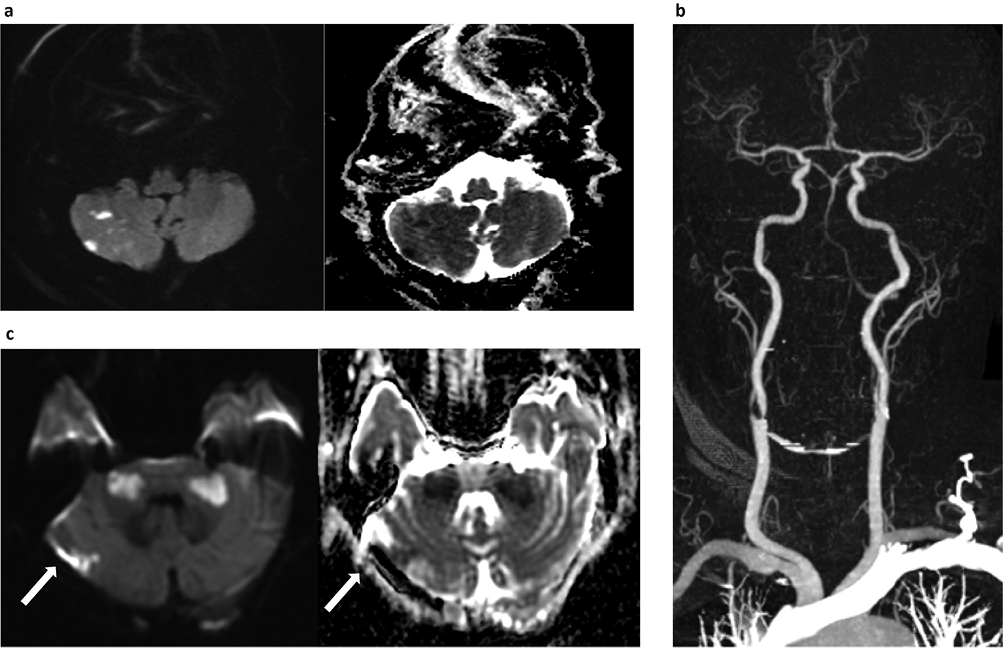
Investigations revealed a mild normocytic anaemia with marked elevation of the erythrocyte sedimentation rate (ESR; 120 mm/hour) and C-reactive protein (129 mg/l). An initial magnetic resonance (MR) scan of the brain demonstrated several areas of restricted diffusion in both cerebellar hemispheres (Figure 1a), consistent with multiple infarcts. He was treated acutely with 300 mg aspirin, followed by 75 mg clopidogrel. A septic screen and transthoracic echocardiogram were normal. Paraneoplastic antibodies and whole-body CT showed no evidence of malignancy. CT angiography confirmed bilateral vertebral artery occlusions (BVAO) and marked narrowing of the right external carotid artery immediately distal to its origin. Atherosclerotic disease with calcification was noted at the origins of the internal carotid arteries bilaterally, causing severe stenosis of the right internal carotid artery (Figure 1b). A 3-day course of intravenous methylprednisolone (1 g/day) was started for a clinically diagnosed GCA, along with dual antiplatelet therapy in the form of aspirin and clopidogrel.
Figure 1 (a) Diffusion-weighted images with corresponding apparent diffusion coefficient maps demonstrating multiple foci of diffusion restriction, consistent with infarcts. (b) Maximum intensity projection image of CT angiogram of the neck and intracranial arteries. The V1/2 segments of the vertebral arteries are completely occluded. There is some reconstitution of flow in the basilar as well as V3/4 segments. (c) Diffusion-weighted images with corresponding apparent diffusion coefficient map from repeat MRI showing bilateral middle cerebellar peduncle infarcts. Extension of infarct (arrows) in the right cerebellar hemisphere is also seen
Four days after initiation of methylprednisolone, he developed dysarthria, bidirectional horizontal nystagmus, and gait and limb ataxia. He was transferred to the regional neurosciences centre where examination revealed normal visual examination and the superficial temporal arteries remained palpable and nontender. ESR remained elevated (120 mm/hour).
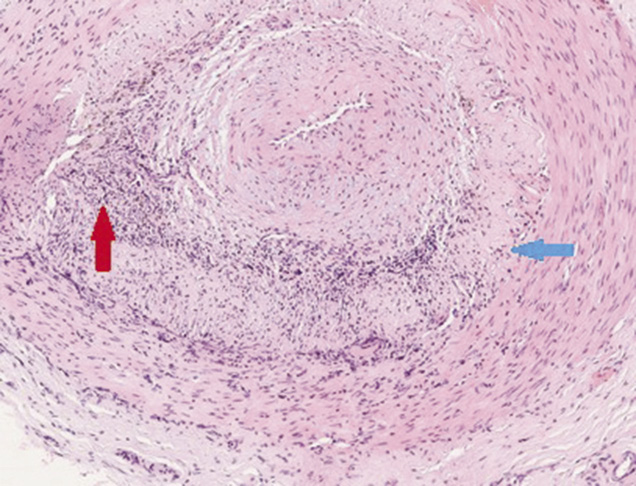
A repeat MR brain demonstrated evolving symmetrical high T2 signal middle cerebellar peduncle (MCP) lesions with restricted diffusion in keeping with new infarcts, along with infarct extension in the right cerebellar hemisphere (Figure 1c). Intravenous methylprednisolone was repeated and heparin was started. A right temporal artery biopsy was performed. This demonstrated inflammatory infiltrate in the tunica media and intima associated with endothelial angiomesodermal hypoplasia, giant multinucleate cells and disruption of the internal elastic lamina, consistent with GCA (Figure 2).
Figure 2 Histology from temporal artery biopsy demonstrating inflammatory infiltrate in the tunica media and intima associated with endothelial angiomesodermal hypoplasia (red arrow), giant multinucleate cells and disruption of the internal elastic lamina (blue arrow) consistent with giant cell arteritis (haematoxylin and eosin-stained section)
Worsening dysarthria and limb ataxia with persistently elevated ESR (49 mm/hour) prompted initiation of cyclophosphamide (six cycles). This was started 2 weeks after the first dose of intravenous methylprednisolone. Heparin was subsequently stopped. Three months after cyclophosphamide, ESR was within normal limits (3 mm/hour). Repeat MR imagery demonstrated maturation of cerebellar infarcts with persistent occlusion of the aforementioned cerebral vessels. Eventually he was able to taper off his steroids completely without any evidence of relapse in consecutive second year. On review, the patient was functionally independent with near-normal speech.
Discussion
This patient presented with clinical features of GCA and developed vertebrobasilar ischaemia despite high-dose oral corticosteroids. His pattern of infarction was unusual with symmetrical MCP involvement. Furthermore, he deteriorated despite high-dose methylprednisolone and heparin, but apparently improved after treatment with cyclophosphamide. Clinical deterioration despite corticosteroid treatment has been discussed in other cases, such as Staunton et al.,2 and can be seen up to 12 days following corticosteroid administration.
BVAO are a rare complication of GCA that carry a high mortality rate (75–80%).3,4 The intracranial vertebral arteries distal to the first 5 mm following penetration of the dura are typically spared, possibly owing to the relative scarcity of elastic tissue in the tunica media and adventitia of the distal intracranial arteries.4 Arteriosclerotic disease is often a contributory factor and has a predilection for the proximal V1 and intracranial V4 segments. In GCA, the V3 and proximal V4 segments are more commonly involved.4
Symmetrical MCP pathology is unusual and the different causes are summarised in Table 1. The MCPs are predominantly supplied by the anterior inferior cerebellar arteries (AICA), but also the superior cerebellar arteries that anastomose with terminal branches of the AICA. We presume that BVAO in this patient resulted in watershed infarction of the MCPs. The imaging features were consistent with infarcts rather than nonischaemic inflammatory lesions.
Table 1 Differential diagnosis of bilateral hyperintensity of the middle cerebellar peduncles
|
Neurodegenerative diseases
|
Multiple systemic atrophy
Spinocerebellar atrophy 2 and 6
|
|
Metabolic diseases
|
Adrenoleukodystrophy
Wilson disease
Alcoholic liver cirrhosis
Hypoglycaemic coma
|
|
Neoplasms
|
Primary CNS lymphoma
Brainstem glioma
Leptomeningeal carcinomatosis
|
|
Cerebrovascular disease
|
AICA infarction
Hypertensive encephalopathy
Pontine infarct with Wallerian degeneration
|
|
Inflammatory and demyelinating disease
|
Multiple sclerosis
ADEM
Behcet disease
Erdheim–Chester disease
HIV encephalopathy
|
ADEM: acute disseminated encephalomyelitis; AICA: anterior inferior cerebellar arteries; CNS: central nervous system
The American College of Rheumatology 1990 diagnostic criteria for GCA are summarised in Box 1.5 More than 90% of patients have elevated acute phase reactants e.g. C-reactive protein and ESR. Thrombocytosis, normocytic anaemia and liver enzyme abnormalities may also be seen.1 The gold standard of diagnosis remains a temporal artery biopsy. The sensitivity of this test ranges from 70% to >90%, therefore, a negative test does exclude the diagnosis.1 Importantly, steroid treatment for 1–2 weeks has little effect on the sensitivity of this test, therefore, empirical treatment in suspected cases need not be delayed.1 Noninvasive diagnosis can also be achieved with colour Doppler and duplex ultrasonography. The detection of a unilateral ‘halo sign’ in the temporal artery by an experienced operator has a specificity of 88–94% and a sensitivity of 61–74%, and would seem a logical first step if locally available.6 18F-fluorodeoxyglucose PET is more sensitive than angiography for the detection of large-vessel GCA (e.g. aortic involvement), with a sensitivity of 63–91% and specificity of 78–94%.7 However, its utility is limited when assessing the temporal and cranial arteries specifically.
Box 1 American College of Rheumatology giant cell arteritis (GCA) diagnostic criteria 19905 – for a diagnosis of GCA to be made, three of five parameters must be fulfilled, yielding a sensitivity of 93% and specificity of 91% compared to other vasculitides
- Age of onset ≥50 years
- New headache
- Temporal artery abnormality e.g. tender palpation or reduced pulsation
- Erythrocyte sedimentation rate ≥50 mm/hour
- Abnormal artery biopsy – vasculitis with mononuclear cell or granulomatous inflammation, often with giant cells
|
Early treatment with high-dose oral corticosteroids is the cornerstone of treatment to prevent cranial ischaemic complications, such as arteritic anterior ischaemic optic neuropathy. An induction pulse of methylprednisone allows for more rapid tapering of oral prednisone.8 In some cases corticosteroids may not prevent GCA-related ischaemia.9,10 The reasons for this are unclear, but both deleterious effects and suboptimal dosing of steroids have been proposed as possible explanations. Evidence for treatment measures in this situation are limited. Cyclophosphamide and tocilizumab have been used for GCA refractory to conventional therapy (high steroid requirement ± first-line immunosuppressant e.g. azathioprine or methotrexate), and the latter has been shown to be superior to placebo in achieving glucocorticoid-free remission over a 26-week or 52-week prednisone tapering period.11,12 Tocilizumab is now approved by The National Institute for Health and Care Excellence (NICE) as a treatment option in relapsing or refractory GCA.13 Once remission is achieved the dose is typically reduced slowly over a period of 1–2 years. For steroid-dependent GCA, alternative immunosuppressants have been studied with variable benefit, including methotrexate, azathioprine, mycophenolate mofetil, etanercept and tocilizumab.13–16
The rational for anticoagulation is based on the principle that granulomatous arteritis may lead to thrombus formation and factors, such as von Willibrand factor, are elevated in GCA.17 Buono et al.9 published a case in which a patient’s visual loss progressed despite high-dose corticosteroid treatment, and an improvement was noted on commencing intravenous heparin; however, prospective evidence for the routine use of anticoagulation are lacking. Two retrospective analyses (n = 166 and n = 143) support the addition of an antiplatelet agent to high-dose corticosteroids in reducing cranial ischaemic complications, thus aspirin or clopidogrel are an important early consideration in suspected GCA.18,19
Conclusion
This case is of interest owing to the rare complication, the unusually aggressive course and the radiological features of what is an otherwise common primary vasculitis. Our patient presented with an acute complication of GCA, with classical features of vasculitis only apparent on thorough review of the history. Vertebrobasilar strokes are a rare manifestation of GCA and bilateral infarction of the MCPs is also noteworthy. The progression of infarcts despite empirical management led to alternative treatment trials, in the form of cyclophosphamide. This appeared to halt the progressive course of the condition and the patient recovered well. The extended spectrum of treatment options in refractory GCA is discussed here, along with the NICE-approved treatment of tocilizumab.
Take home messages
- GCA should be considered in patients aged over 50 years old presenting with posterior circulation stroke and raised inflammatory markers.
- Empirical treatment with high-dose steroids and antiplatelet therapy should be considered in suspected cases.
- Our case highlights the importance of considering aggressive immunosuppression to prevent further ischaemic episodes.
- The symmetric MCP lesions can be due to watershed hypoperfusion from symmetrical vertebrobasilar involvement.
- GCA has a predilection for the vertebrobasilar system and symmetric involvement is unusual in embolic disease.
References
1 Borchers AT, Gershwin ME. Giant cell arteritis: a review of classification, pathophysiology, geoepidemiology and treatment. Autoimmun Rev 2012; 11: A544–54.
2 Staunton G, Stafford F, Leader M et al. Deterioration of giant cell arteritis with corticosteroid therapy. Arch Neurol 2000; 57: 581–4.
3 Rüegg S, Engelter S, Jeanneret C et al. Bilateral vertebral artery occlusion resulting from giant cell arteritis: report of 3 cases and review of the literature. Medicine (Baltimore) 2003; 82: 1–344.
4 Wilkinson IMS, Russell RWR. Arteries of the head and neck in giant cell arteritis: a pathological study to show the pattern of arterial involvement. Arch Neurol 1972; 27: 378–91.
5 Hunder GG, Bloch DA, Michel BA et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33: 1122–8.
6 Arida A, Kyprianou M, Kanakis M et al. The diagnostic value of ultrasonography-derived edema of the temporal artery wall in giant cell arteritis: a second meta-analysis. BMC Musculoskelet Disord 2010; 11: 44.
7 Besson FL, Parienti J-J, Bienvenu B et al. Diagnostic performance of 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a systematic review and meta-analysis. Eur J Nucl Med Mol Imaging 2011; 38: 1764–72.
8 Hunder GG, Sheps SG, Allen GL et al. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med 1975; 82: 613–8.
9 Buono LM, Foroozan R, de Virgiliis M et al. Heparin therapy in giant cell arteritis. Br J Ophthalmol 2004; 88: 298–301.
10 Loock J, Henes J, Kötter I et al. Treatment of refractory giant cell arteritis with cyclophosphamide: a retrospective analysis of 35 patients from three centres. Clin Exp Rheumatol 2012; 30: S70–6.
11 National Institute for Health and Care Excellence (2018). Tocilizumab for treating giant cell arteritis. https://www.nice.org.uk/guidance/ta518 (accessed 07/11/18).
12 Hoffman GS, Cid MC, Hellmann DB et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002; 46: 1309–18.
13 Unizony S, Arias-Urdaneta L, Miloslavsky E et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res (Hoboken) 2012; 64: 1720–9.
14 De Silva M, Hazleman BL. Azathioprine in giant cell arteritis/polymyalgia rheumatica: a double-blind study. Ann Rheum Dis 1986; 45: 136–8.
15 Sciascia S, Piras D, Baldovino S et al. Mycophenolate mofetil as steroid-sparing treatment for elderly patients with giant cell arteritis: report of three cases. Aging Clin Exp Res 2013; 31; 24: 273–7.
16 Martínez-Taboada VM, Rodríguez-Valverde V, Carreño L et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis 2008; 67: 625–30.
17 Nordborg E, Andersson R, Tengborn L et al. von Willebrand factor antigen and plasminogen activator inhibitor in giant cell arteritis. Ann Rheum Dis 1991; 50: 316–20.
18 Nesher G, Berkun Y, Mates M et al. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum 2004; 50: 1332–7.
19 Lee MS, Smith SD, Galor A et al. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum 2006; 54: 3306–9.