Introduction
Pulmonary hypertension (PH) is present when the mean pulmonary arterial pressure (mPAP) is ≥25 mmHg measured during right heart catheterisation (RHC).1 This is a relatively arbitrary cut-off, originally chosen to prevent overdiagnosis and treatment. In 2018, a revised threshold of 20 mmHg was proposed to define PH. This takes into account a systematic review of mPAP ranges for healthy individuals, where ≥20 mmHg corresponds with greater than two standard deviations above the population mean.2
Clinical classification
PH is grouped into five clinical groups with similar pathophysiology, haemodynamics and response to treatment within each group. These are summarised in Table 1.
Table 1 Clinical and haemodynamic classification of pulmonary hypertension2
|
|
|
|
|
|
Group I
Pulmonary arterial hypertension (PAH)
|
Idiopathic (IPAH)
Heritable (HPAH)
Drug and toxin-induced
Connective tissue disease (CTD–PAH)
Portal hypertension (Portopulmonary, PoPAH)
Congenital heart disease (CHD–PAH)
Pulmonary veno-occlusive disease (PVOD)
Others: HIV, schistosomiasis, persistent PH of the newborn
|
≥3
|
≤15
|
|
Group II
Left heart disease (PH–LHD)
|
Heart failure with preserved left ventricular ejection fraction (HFpEF)
Heart failure with reduced left ventricular ejection fraction (HFrEF)
Valvular heart disease
|
Isolated postcapillary
|
≤3
|
>15
|
|
Combined pre- and postcapillary
|
≥3
|
>15
|
| |
|
|
|
Group III
Lung disease and/or hypoxia
|
Restrictive lung disease
Obstructive lung disease
Hypoxia without lung disease
|
|
≥3
|
≤15
|
|
Group IV
Pulmonary arterial obstruction
|
Chronic thromboembolic pulmonary hypertension (CTEPH)
Other pulmonary artery obstructions
|
|
≥3
|
≤15
|
|
Group V
Multifactorial or unclear causes
|
Haematological disorders
Systemic and metabolic disorders
Complex congenital heart disease
|
|
≥3
|
≤15
|
| |
|
|
|
|
As shown in Case 1, pulmonary arterial hypertension (PAH, Group I) is a primary pulmonary vasculopathy where increases in pulmonary vascular resistance (PVR) lead to progressive right ventricular strain and failure.3 PH due to left heart disease (LHD, Group II) is defined at RHC by a pulmonary arterial wedge pressure (PAWP) >15 mmHg and arises from an increase in left atrial pressure due to underlying LHD.4 PH due to lung disease and/or hypoxia (Group III) occurs due to mechanisms such as hypoxic vasoconstriction and obliteration of the pulmonary vascular bed.5 PH secondary to pulmonary artery obstruction (Group IV) primarily consists of chronic thromboembolic pulmonary hypertension (CTEPH). This is caused by nonresolution and organisation of thromboembolic material following pulmonary embolism (PE) and is notable for the potentially curative surgical treatment options available.6 Group V comprises a mix of diseases with multifactorial or unclear mechanisms.2
PH may also be defined by the location of the disease process in relation to the pulmonary vascular capillary bed where Groups I, III, IV and V typically occur precapillary.2 Groups II and V can exist as isolated postcapillary (IpcPH) where there is no intrinsic elevation in PVR, or as combined post- and precapillary (CpcPH) where the PVR is raised.4
Epidemiology
Ninety-six per cent of PH in Europe is due to left heart or chronic lung disease.7 In the UK, PAH affects approximately 48–55 people per million (ppm) with an incidence of 6 ppm per year. The incidence of CTEPH is acknowledged to be under-reported at 3–6 ppm per year with a prevalence of 26–38 ppm.8–10 Globally, the causes of PH can differ significantly, with cases of PH related to sickle cell disease and infectious diseases (HIV, schistosomiasis, post-Streptococcus rheumatic heart disease) more prevalent in countries where these are still endemic.7
In the UK, the mean age at diagnosis of idiopathic pulmonary arterial hypertension (IPAH) is 60 years old, and 37.7% of IPAH patients are male.11 Over the last 20 years, newly diagnosed patients with PAH are more likely to be older and have multiple comorbidities, reflecting the increased recognition of PH in older patients and their onward referral.12,13
Prognosis
Whilst PH remains a progressive and life-limiting condition, prognosis significantly varies depending on aetiology and available treatments.11 In the UK, five-year survival for Group II is 45% and Group III is 23%. However, five-year survival rates for younger patients (18–53 years old) with PAH have improved over the last decade from 74% to 83–85%, and patients with CTEPH who undergo surgical treatment have a five-year survival of 85%.11,12 In PAH patients who respond to vasoreactivity challenge and are treated with calcium channel blockers, PH might not limit their life expectancy.
Pathophysiology
In patients with PAH, remodelling and obstruction of the pulmonary vasculature leads to increased PVR. As the right ventricle (RV) afterload increases, right heart strain develops with pressure overload, causing the RV to remodel from a low to a high-pressure system. In response to the subsequent volume overload, the RV dilates in an attempt to maintain stroke volume, and functional tricuspid regurgitation (TR) develops. Eventually the RV will decompensate leading to the development of right heart failure.14,15
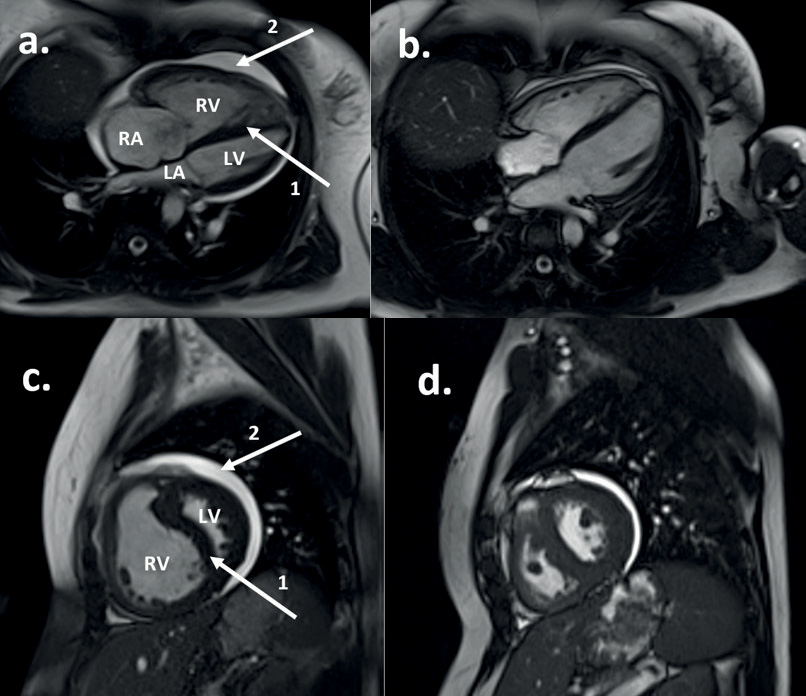
Figure 1(a,c) demonstrates features of RV decompensation on a cardiac magnetic resonance image.
Figure 1 Cardiac magnetic resonance image of a patient with IPAH (see Case 1), before (a and c) and after (b and d) treatment with disease-targeted therapy. Panels (a) and (b) demonstrate the four-chamber transverse plane; (c) and (d) demonstrate the two-chamber sagittal plane. Panels (a) and (c) show the heart in end-diastole with an enlarged RV causing compression of the LV with septal deviation (arrow 1). Arrow 2 denotes a pericardial effusion. Panels (b) and (d) demonstrate end-diastolic views in the same patient after treatment, indicating an overall reduction in cardiac size, improvement in the pericardial effusion and normalisation in the RV size.
Case 1
A 35-year-old female presented after an episode of presyncope following the use of cyclizine for nausea. On further questioning she had a one-year history of exertional breathlessness and had experienced two recent syncopal episodes. Transthoracic echocardiogram (TTE), which demonstrated a raised systolic pulmonary artery pressure (sPAP) of 134 mmHg and a dilated and impaired RV. Computerised tomography pulmonary angiography (CTPA) reported as demonstrating bilateral pulmonary emboli with right heart dilation. She was treated with warfarin and subsequently discharged. Despite anticoagulation, her symptoms progressed and she developed exertional chest pain and worsening presyncope, prompting re-admission and transfer to the Scottish Pulmonary Vascular Unit (SPVU) with a provisional diagnosis of CTEPH. Spirometry was preserved, DLCO (transfer factor for carbon monoxide) was reduced at 53% predicted, NTproBNP was not elevated. She walked 426 m on a six-minute walk test (6MWT) and mildly desaturated to an SpO2 of 94%. RHC revealed a raised mPAP of 71 mmHg, a normal PAWP of 9 mmHg and a raised PVR of 17.2 Wood units (WU). Radiological review of the initial CTPA did not agree with the diagnosis of acute PE, and the pulmonary angiogram, magnetic resonance pulmonary angiogram (MRPA) and repeat CTPA also did not demonstrate any features of chronic or acute pulmonary thromboembolic disease. Due to an absence of an underlying aetiology, her diagnosis was revised to IPAH. Due to the history of syncopal episodes, she was commenced on triple disease-targeted therapy with IV Epoprostenol, Sildenafil and Macitentan. She had significant improvement in her symptoms afterwards, has returned to work and is now World Health Organization functional class (WHO FC) I. After six years of treatment with IV Epoprostenol, she developed a Hickman line infection, prompting review of her therapies. The IV Epoprostenol was replaced with oral Selexipag without evidence of deterioration (Figures 1 and 2).
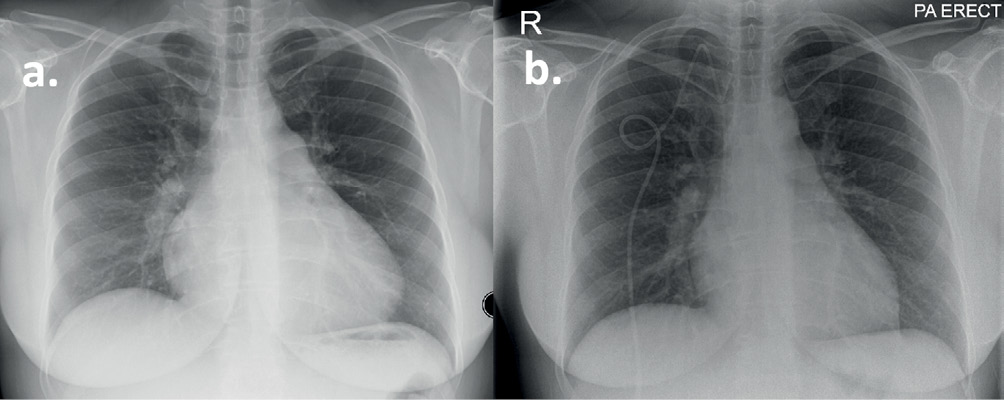
Figure 2 Chest X-ray of a patient with IPAH (see Case 1). (a) Postero-anterior projection at diagnosis demonstrating cardiomegaly and dilated pulmonary arteries. (b) Following six years of treatment with triple disease-targeted therapy demonstrating a right-sided central venous tunnelled catheter for administration of Epoprostenol and improvement in cardiac size.
Clinical assessment
It remains the case that patients with PH are diagnosed late into their disease course (Box 1).16–18 A baseline knowledge of the condition and an index of suspicion are required to facilitate diagnosis. Table 2 shows how patients with PH may present to a variety of secondary care specialties.
Box 1 Why is there a delay in the diagnosis of PH?
- Nonspecific early symptoms and signs
- Symptoms and signs are attributed to more common comorbidities
- Rare condition and therefore a low index of suspicion
- Complex route of presentation that may involve cardiology, respiratory or other specialties
- Lack of awareness of referral pathways, guidelines and tertiary centres
- No effective screening strategies in majority of conditions that cause PH, e.g. IPAH, chronic heart disease, liver disease
- Screening not performed, e.g. postpulmonary embolism, connective tissue disease, genetic screening
Table 2 Aetiology of PH according to medical specialty
|
|
|
|
Respiratory
|
Any form of PH including Group III
|
|
Cardiology
|
Any form of PH including Group II, permanent pacemaker (relationship to pulmonary emboli)
|
|
Acute Medicine
|
Following or associated with acute venous thromboembolism
|
|
Gastroenterology
|
Portopulmonary PH
|
|
Neurology
|
Interferon related PH in multiple sclerosis, ventriculoatrial shunts (PE)
|
|
Anaesthetics
|
Any form of PH – unexpected hypotension or hypoxaemia during surgery
|
|
Rheumatology
|
CTD–PAH (scleroderma, systemic lupus erythematosus, mixed connective tissue disease, sarcoidosis)
|
|
Genetics
|
Hereditary haemorrhagic telangiectasia
|
|
Ear, Nose and Throat
|
Hereditary haemorrhagic telangiectasia, vocal cord palsy
|
|
Haematology
|
Myeloproliferative disorders, POEMS syndrome, tyrosine kinase inhibitors
|
|
Dermatology
|
Hereditary haemorrhagic telangiectasia
|
|
Infectious Diseases
|
HIV, schistosomiasis, recreational drug use
|
|
Psychiatry
|
Drug-induced PH due to anorexigens, and serotonin reuptake inhibitors
|
|
Oncologists
|
Tumour emboli, drug-induced PH due to immunotherapy, tyrosine kinase inhibitors and chemotherapy
|
| |
|
PH progresses at different rates depending on aetiology, with conditions such as pulmonary veno-occlusive disease (PVOD) and connective tissue disorder–pulmonary arterial hypertension (CTD–PAH) following a more aggressive course.11 Earlier signs and symptoms are subtle, nonspecific and easily missed. Commonly reported symptoms are related to RV dysfunction and include dyspnoea, fatigue, chest pain, exertional presyncope and reduced exercise tolerance.19 Abnormalities of the pulmonary vasculature or secondary bronchial artery hypertrophy may cause haemoptysis, and compressive pathology due to enlargement of the pulmonary artery may cause coronary ischaemia or left vocal cord palsy.19 Clinical signs of right heart strain may be absent, but when present include an accentuated component of the second heart sound over the pulmonary valve (loud P2), a pansystolic murmur of TR, a diastolic murmur of pulmonary regurgitation and an RV heave. Signs of right heart failure occur later in the disease and include peripheral oedema, ascites, hepatomegaly and a raised jugular venous pulse.19
A history should assess for possible underlying aetiologies and the disease severity. This should include past medical conditions that predispose to PH (e.g. connective tissue disease, previous PE or liver disease), a family history (relevant for conditions such as hereditary haemorrhagic telangiectasia, PVOD, heritable pulmonary arterial hypertension [HPAH] and CTD–PAH), and risk features for cardiorespiratory disease. Patients are stratified according to WHO FC from I–IV, which allows a simple measure of disease severity.19
PH is not a condition commonly diagnosed in primary care. The classical symptom of exertional dyspnoea should prompt onward referral to cardiology or respiratory specialists for further investigation. This should be considered in patients who do not respond to first-line treatments, who have risk factors for PH or where the diagnosis is not clear.
Investigations
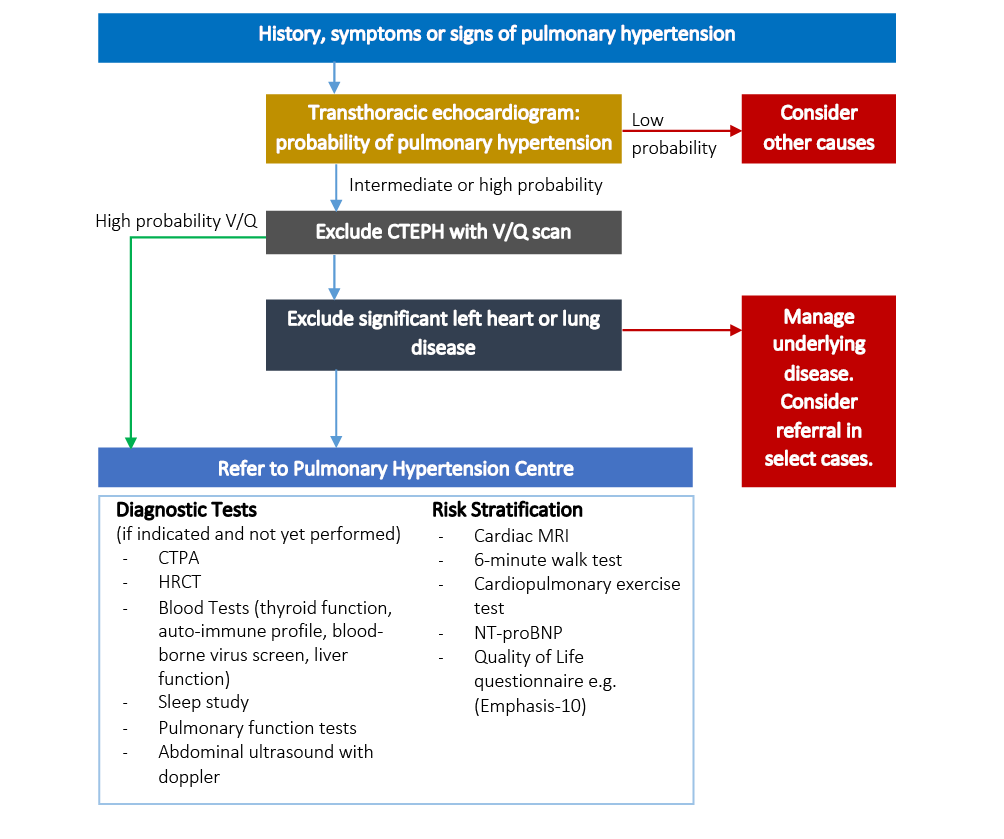
Baseline investigations should diagnose the presence of PH, assess causes and risk stratify patients (see Figure 3). Transthoracic echocardiography remains the keystone noninvasive screening test and should be used to assign a probability of PH.19 Using the simplified Bernoulli equation, the tricuspid regurgitation pressure gradient (TRPG) is calculated from the peak TR velocity and is added to an estimation of right atrial pressure to calculate the sPAP. TRPG ≤31 mmHg suggests a low probability of PH, whilst ≥46 mmHg gives a high probability. TRPG estimates of 32–45 give an intermediate probability, and patients in this category, or where a TR is not measurable, should be assessed in conjunction with other echocardiographic parameters of PH (Box 2).19,20 TTE may allow a quantification of the severity of PH by assessing the RV function and size using metrics such as a tricuspid annular plane systolic exertion of <15 mm and a left ventricle (LV) eccentricity index in diastole of >1.7.21 The DETECT and ASIG algorithms have been proposed as screening mechanisms for patients with systemic sclerosis, using a combination of routine investigations to guide the requirement for TTE.22
Figure 3 Diagnostic algorithm for PH
Box 2 Other echocardiographic signs of pulmonary hypertension
A TRPG in the range of 32–45 mmHg with signs from two of the categories below (A/B/C) should prompt further investigation and referral.
- (A) Ventricles: RV:LV basal diameter >1.0, flattening of the intraventricular septum
- (B) Pulmonary artery: pulmonary valve acceleration time <105 ms, pulmonary artery diameter >25 mm, early diastolic pulmonary regurgitation velocity >2.2 m/s
- (C) Inferior vena cava and right atrium: decreased inspiratory collapse of inferior vena cava, enlarged right atrium
Table 3 demonstrates how common testing modalities may help in the diagnosis of PH secondary to LHD or chronic lung disease. The diagnosis of PH–LHD, especially heart failure with preserved ejection fraction, can be challenging. It is important to differentiate this group as further investigations may help to refine the diagnosis but do not lead to subsequent management options. Multiple studies have shown no benefit or deterioration amongst Group II patients treated with pulmonary artery vasodilators.4 PH–LHD patients tend to be older (>70 years), have comorbidities (diabetes, systemic hypertension, dyslipidaemia, atrial fibrillation) which increases cardiovascular risk and may have undergone previous cardiac intervention such as valve replacement or coronary angiography.4 Pretest probability scoring systems such as the H2FPEF and OPTICS score allow further quantification.23,24 Patients with PH–LHD should be managed according to the underlying disease. Patients with PH due to lung disease and/or hypoxia may still be considered for tertiary referral if the extent of pulmonary hypertension is felt disproportionate to the disease process because these patients may benefit from a trial of pulmonary vasodilator therapy. Features favouring proportional PH include moderate or severe impairment on pulmonary function testing, characteristic lung disease features on computerised tomography (CT) imaging, mild to moderate PH at RHC and ventilatory limitation on cardiopulmonary exercise testing.5
Table 3 Investigations in PH and how they may aid differentiation of aetiology
|
|
|
|
|
Electrocardiogram
|
Right heart strain
|
Atrial fibrillation, left bundle branch block, left axis deviation, left ventricular hypertrophy
|
|
Chest X-ray
|
Enlarged pulmonary arteries, cardiomegaly
|
Features of pulmonary oedema, chronic lung disease
|
|
Arterial blood gas analysis
|
Low PaCO2
|
Type 2 respiratory failure may indicate chronic lung disease
|
|
CT imaging
|
Features of CTEPH (web disease, pulmonary arterial tree occlusion, mocasism, bronchial artery hypertrophy, calcified thrombus)
|
Features of left heart disease (dilated left atrium)
|
|
Features of PVOD (septal lines, mediastinal lymphadenopathy, centrilobular ground glass opacities, pleural effusions)
|
Characteristic features of airway or parenchymal abnormalities
|
|
Transthoracic echocardiogram
|
TRPG ≥46
TRPG 32–45 with other parameters of PH
|
Features of left heart disease: dilated left atrium, left ventricular systolic dysfunction, left-sided valvular heart disease, raised E/e ratio.
|
|
Cardiopulmonary exercise testing
|
Exercise capacity limited by oxygen transport abnormality
|
Exercise capacity limited by combination of reduced ventilatory capacity and increased ventilatory demand
|
|
Abnormal gas exchange with raised ventilatory equivalents, PetCO2, high dead space, wide alveolar–arterial gradient and desaturation on exertion
|
| |
|
|
Patients with a high probability, or intermediate probability with other features of PH, on TTE should be referred to a tertiary PH centre for consideration of further assessment. In the UK, PH care is provided through a network of eight centres, and in Scotland this is managed by the SPVU. Patients with Group II and Group III disease should be considered for referral if the extent of PH is disproportionate to the underlying disease.4,5 Patients with severe symptoms (presyncope or syncope) or in right heart failure at presentation should be fast tracked.20
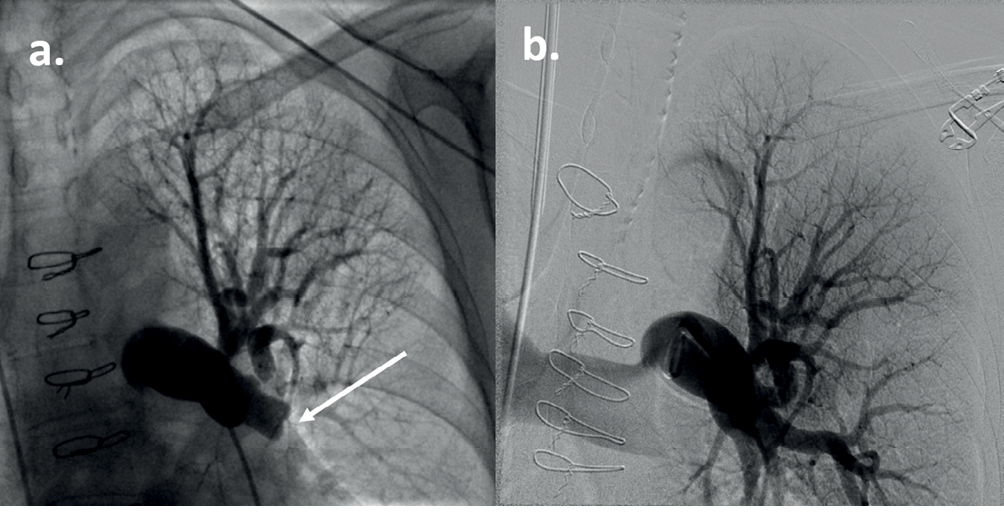
All patients in whom precapillary PH is suspected should undergo RHC. At RHC, the Swan–Ganz catheter measures the intracardiac and pulmonary artery pressures, cardiac output and mixed venous saturations in the main pulmonary artery.25 Further provocatory testing may be performed to refine the diagnosis, including fluid challenge and exercise testing. Vasoreactivity testing with nitric oxide is performed in patients with IPAH and HPAH to assess vasodilator response which may identify them as patients who can be treated successfully long term with calcium channel blockers. In cases of suspected CTEPH, conventional pulmonary angiography can be performed (Figure 4). In cases of a suspected left to right shunt, a saturation run is performed, where oxygen saturation of blood is measured in the inferior and superior vena cava, right heart and pulmonary artery to isolate the source.
Figure 4 Conventional pulmonary angiogram of the left upper thorax in a patient with CTEPH. (a) The arrow indicates a complete occlusion of the left lower lobe pulmonary artery. Sternotomy wires are present from a previous repair of a ventricular septal defect. (b) Pulmonary angiogram with digital subtraction angiography in the same patient following pulmonary endarterectomy. There is revascularisation of the left lower lobe pulmonary artery.
As patients with HPAH, most commonly with a monoallelic mutation in the BMPR2 gene, present at a younger age with a worse prognosis,26 genetic screening should be considered in all patients with IPAH, even in the absence of a family history. Twenty-five per cent of IPAH patients have a BMPR2 mutation and this opens up the possibility of genetic screening for family members if positive,26,27 as shown in Case 2. Patients with features of PVOD may be homozygote for mutations in the EIF2AK4 gene, which may enable diagnostic confirmation of this condition.26
Case 2
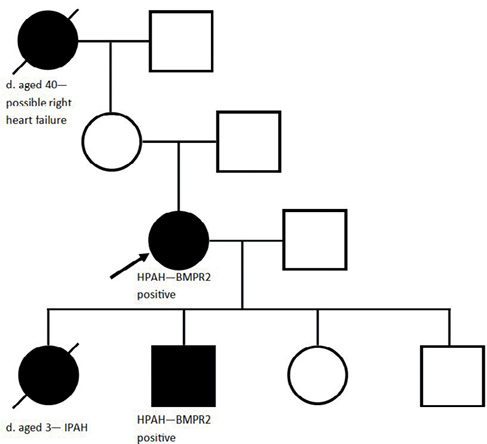
A 41-year-old female and current smoker was referred to SPVU with a two-year history of gradually worsening exertional breathlessness, in keeping with WHO FC III. Her grandmother had died aged 40 with ‘dropsy’ and her daughter had died aged three years following a short-lived illness that was subsequently diagnosed at postmortem as IPAH. On examination, the patient had a loud P2, an RV heave, was centrally cyanosed and systemically hypotensive at rest (93/67 mmHg). NTproBNP was not elevated and she walked 331 m on a 6MWT, mildly desaturating to an SpO2 of 94%. TTE demonstrated a TRPG of 55 mmHg and a severely dilated RV. CT imaging demonstrated main pulmonary artery and RV dilation, mild emphysema and no features of thrombotic disease. RHC demonstrated a raised mPAP of 59 mmHg, a normal PAWP of 8 mmHg and a very high PVR of 22.2 WU. With the positive family history, these results were in keeping with a diagnosis of HPAH. There were concerns about treating with a phosphodiesterase-5 inhibitor due a family history of retinitis pigmentosa, and hence treatment with Bosentan was commenced. Riociguat was added six years after diagnosis. She remains stable on these ten years after diagnosis and is now WHO FC II. Given the family history, her remaining three children were screened using TTE. Her 14-year-old son had a raised TRPG of 61 mmHg and went on to have HPAH confirmed at RHC. He remains well in FC II on Bosentan monotherapy. Subsequent genetic analysis revealed the patient and her son were positive for a monoallelic mutation in the BMPR2 gene (Figure 5).
Figure 5 Genetic pedigree of a female patient with HPAH (see Case 2). The black arrow indicates the index patient. A filled black box indicates an individual affected with PH and a diagonal line indicates an individual is deceased (d. indicates age at death).
Management
Early combination drug therapy has been shown to improve outcomes in PAH.28 Current pharmacological therapies target three distinct biological pathways: the nitric oxide, endothelin and prostanoid pathways, which is further elaborated in Table 4.29,30 Current guidelines advocate initial combination therapy unless contraindicated.30 For patients with more severe disease or who do not improve following initial treatment, parenteral treatment may be required, such as with Epoprostenol via a tunnelled central venous catheter.29 A subset of IPAH will show haemodynamic improvement with pulmonary vasoreactivity testing during RHC and can be treated with calcium channel blockers, often with dramatic clinical improvement.19
Table 4 Therapeutic targets in pulmonary hypertension
|
|
|
|
|
|
|
|
|
|
Sildenafil
|
Oral, intravenous
|
Three times daily – variable dosing*
|
Indigestion, heartburn, headache, flushing, jaw pain, hypotension, visual impairment, hearing impairment, priapism
|
|
Tadalafil
|
Oral
|
Once daily – 20 mg, 40 mg
|
|
|
|
Riociguat
|
Oral
|
1–2.5 mg three times a day
|
Hypotension
Contraindicated with PDE5i
|
|
|
Bosentan
|
Oral
|
62.5–125 mg twice daily
|
Liver function test derangement and anaemia (requires blood monitoring)
Teratogenic
Headache, flushing, hypotension, fluid retention
|
|
Macitentan
|
|
10 mg once daily
|
|
Ambrisentan
|
|
5–10 mg once daily
|
|
|
Iloprost
|
Nebulised
Intravenous
|
10–20 μg seven times a day
Continuous infusion
|
Difficulty with nebuliser
Flushing, trismus, hypotension
|
|
Selexipag
|
Oral
|
200–1,600 μg twice daily
|
Flushing, diarrhoea, nausea, vomiting
|
|
Treprostenil
|
Subcutaneous, intravenous
|
|
Complications from subcutaneous access
|
|
Epoprostenol
|
Intravenous
|
Continuous infusion – starting at 2 ng/kg/min and increased in intervals
|
Complications arising from venous access
Hypotension, flushing, jaw pain, diarrhoea, extremity pain
|
| |
|
|
|
|
Judgement of clinical deterioration and requirement for escalation of therapy is based on risk stratification and includes the patient’s diagnosis, exercise capacity, WHO FC, haemodynamic values, NTproBNP/BNP assay and imaging findings, as expressed in scores such as the REVEAL 2.0 risk calculator or the European Respiratory Society/European Society of Cardiology risk table.30,31
Treatment options for all patients should include optimal diuresis, appropriate vaccination as for any chronic respiratory disease and consideration of long-term oxygen if respiratory failure is present. Beta-blockers and cyclizine should be avoided because they cause a reduction in cardiac output. Nitrates are contraindicated with phosphodiesterase-5 inhibitors and cannot be used for anginal treatment in patients taking these. As shown in Case 3, female PH patients should be counselled about the high mortality associated with pregnancy, and advice given on contraception and termination.32 Conditions that aggravate PH such as dysrhythmias, infection and acute PE, should be detected and treated promptly. Patients with acute RV failure may need to be treated in a high-dependency environment with inotropic and vasopressor support.33 Bilateral lung transplantation is considered in patients who do not respond to, or deteriorate whilst on, optimal therapy.19 Low-intensity aerobic exercise is safe and should be encouraged in patients with PH to reduce deconditioning and improve patients’ quality of life.34 Patients should not be told to avoid exercise, yet should be warned against activities leading to symptoms of presyncope or syncope.
As shown in Case 3, patients with CTEPH should be referred for consideration of surgical treatment.6 In the UK, this is performed at the Royal Papworth Hospital in Cambridge. Surgical options include pulmonary endarterectomy (PEA) for proximal disease and balloon pulmonary angioplasty for more distal disease. Whilst response to PEA can be excellent, it involves hypothermic circulatory arrest and carries a significant perioperative mortality risk of 2–5%.35 CTEPH patients require lifelong anticoagulation.6 There is no strong evidence for anticoagulation in other forms of precapillary PH; however, it is important to note that in CTD–PAH anticoagulation may be harmful due to an increased bleeding risk and should only be commenced when there is a clear indication.36
Case 3
A previously well 24-year-old female presented acutely with a one-week history of exertional breathlessness following a short-haul flight three weeks earlier. CTPA confirmed extensive bilateral pulmonary emboli and she was subsequently discharged on Rivaroxaban. She became pregnant three months later and her anticoagulation was changed to low molecular weight heparin. It was noted during an antenatal visit that she was breathless on exertion (WHO FC II), prompting assessment with TTE which demonstrated a reduced pulmonary valve acceleration time of 73 ms (NR >105 ms), mild right ventricular systolic dysfunction and RV dilation, without a visible TR jet. A ventilation–perfusion (V/Q) scan demonstrated multiple large, mismatched perfusion defects (Figure 6). She was referred to SPVU, at which point her fetus was 12 weeks’ gestation. She walked 470 m on a 6MWT and significantly desaturated to an SpO2 of 80%. She underwent RHC, revealing a raised mPAP of 40 mmHg, normal PAWP of 6 mmHg and raised PVR of 6.4 WU. Neither MRPA nor conventional pulmonary angiogram were performed due to pregnancy. Repeat CTPA demonstrated features of chronic thromboembolic disease (mosaicism, bronchial artery hypertrophy, and web and occlusive disease) in keeping with a diagnosis of CTEPH (Figure 7). She was commenced on medical treatment with Sildenafil and referred to the Royal Papworth Hospital who advised that PEA was to be avoided during pregnancy. The patient was counselled on the risks of pregnancy in PH but elected against termination. Unfortunately, intrauterine death occurred at 26 weeks’ gestation and the patient underwent an uneventful vaginal delivery of fetal products. Tests at this time showed her to be triple positive for antiphospholipid syndrome, and her anticoagulation was changed to warfarin. She underwent PEA at Royal Papworth Hospital 16 months after her initial acute PE, and follow-up RHC six months later confirmed haemodynamic improvement with a normal mPAP of 23 mmHg, PAWP 11 mmHg and PVR 2.0 WU. Sildenafil was discontinued post-surgery. The patient has subsequently had a successful pregnancy and continues on lifelong anticoagulation.
Figure 6 Ventilation and perfusion (V/Q) scan in a patient with CTEPH (see Case 3) demonstrating multiple mismatched perfusion defects.
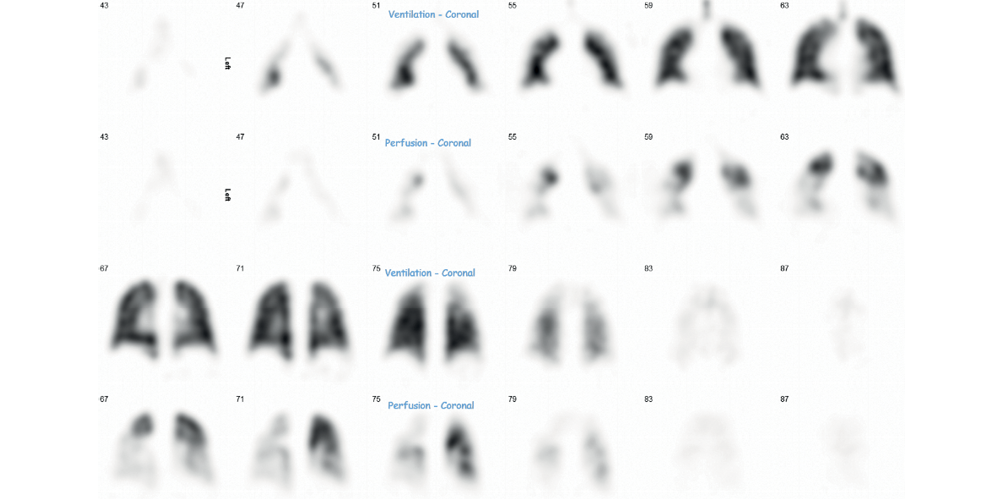
Figure 7 CT pulmonary angiogram of a patient with CTEPH (see Case 3). There is evidence of chronic thromboembolic disease in the pulmonary arteries. The main pulmonary artery is dilated at 34 mm and is wider than the aorta, which is in keeping with PH.
Conclusion
PH is a multifaceted condition resulting from a wide range of underlying conditions. Therapeutic avenues have expanded enormously over the last two decades resulting in major improvements in prognosis for some presentations. As a consequence, detailed investigation is essential to establish the precise aetiology and hence the optimum treatment. Unfortunately, PH is still often diagnosed months or years after the onset of symptoms, and screening strategies are unlikely to improve this in the foreseeable future. Knowledge of the aetiology, presentation and treatment options for this condition is therefore important in many areas of medicine to improve the management of this life-altering condition.