Introduction
Heterozygous mutations in the HNF4A gene are a recognised cause of hyperinsulinaemic hypoglycaemia in infancy, and evolve into diabetes in adulthood associated with insulin deficiency (maturity-onset diabetes of the young [MODY] 1).1
We present the case of a 69-year-old male who was referred with lifelong hypoglycaemia that was found to be due to a pathogenic HNF4A mutation. The patient had not developed diabetes and was successfully treated with diazoxide.
Case presentation
The patient was referred to our endocrinology service in 2017, after moving to the UK from South Africa, with increasingly troublesome episodes of shakiness, disorientation and loss of consciousness that had occurred throughout his life. These episodes were usually resolved by eating carbohydrate-rich foods; however, paramedics were occasionally called and he was found to have capillary glucose levels as low as 2.8 mmol/l. Consistently, his symptoms resolved after oral glucose administration and correction of hypoglycaemia. He was macrosomic at birth (5.90 kg), suffered neonatal hypoglycaemia and was overweight as a small child. He reported that his primary school teacher always gave him a glass of water with sugar when he arrived in school and at the age of 9 years was formally diagnosed with a hypoglycaemic disorder; no cause was identified. His mother did not suffer diabetes in pregnancy; however, both mother and maternal grandmother developed late-onset diabetes responsive to sulphonylurea (Figure 1a). One of his two sons was also macrosomic at birth (4.25 kg), suffered neonatal hypoglycaemia, was diagnosed with diabetes aged 34 years and treated with a sulphonylurea.
Figure 1 a) Family pedigree indicating, where known: birthweight, presence of neonatal hypoglycaemia, diabetes age and BMI at diagnosis, and treatment. b) Ambulatory glucose profile over 7 days. BMI: body mass index at diagnosis; BW: birth weight; Diag: diagnosed with HNF4A mutation; DM: diabetes mellitus; NK: not known; ‘Normal’: described by patient as not under or overweight OHA NOS: oral hypoglycaemic agent not otherwise specified; SU: sulphonylurea.
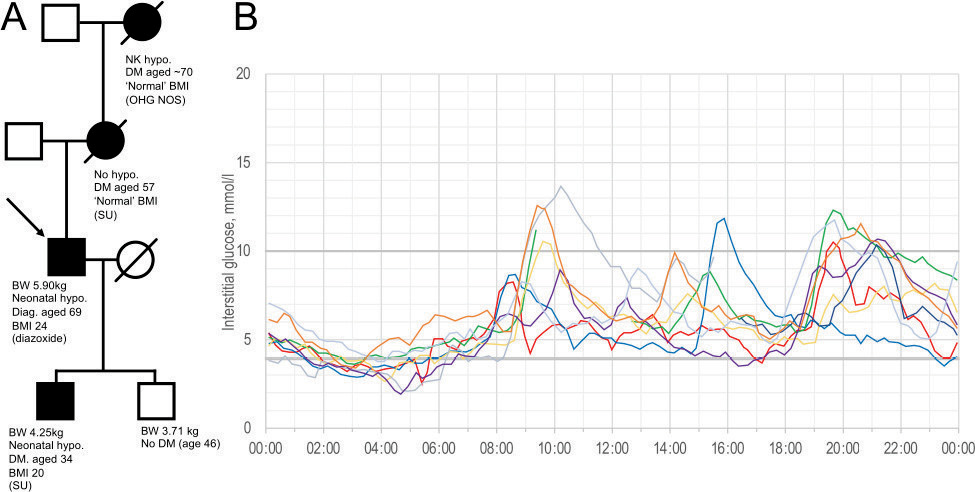
A 7-day continuous subcutaneous blood glucose monitor revealed persistent early morning low glucose and modest post-prandial excursions (Figure 1b). Clinical examination was normal; BMI was 24.0 kg/m2. HbA1c was low (31 mmol/mol), fasting glucose was 3.6 mmol/l, and C-peptide and insulin were and 14 pmol/l and 240 mmol/l, respectively. A genetic panel identified a truncating mutation of HNF4A, c.421C>T p.(Arg141). He was treated successfully with diazoxide, with dose adjustment guided by symptoms and occasional use of a Freestyle Libre flash glucose meter (Abbott, Maidenhead, UK). He has remained free from significant hypoglycaemia since presenting in 2018.
Discussion
Heterozygous mutations resulting in haploinsufficiency of the HNF4A gene are associated with in utero hyperinsulinaemia, macrosomia and neonatal hypoglycaemia. Hypoglycaemia is normally transient with resolution of symptoms expected during the first year.1 One reported individual required diazoxide treatment until aged 12 years,2 while asymptomatic biochemical hypoglycaemia has been reported after 21 years.3 In adults with HNF4A mutations, there is usually a switch to insulin deficiency and MODY, which is highly sensitive to sulphonylurea treatment.4 However, other phenotypes of pathogenic HNF4A mutations have been described including neonatal hypoglycaemia that resolves and does not progress to MODY, and MODY without neonatal hypoglycaemia.4 The variable progression to MODY may be due to eventual burnout of beta cell function or, perhaps more likely, an epigenetic switch in HNF4A transcription.3
The c.421C>T HNF4A mutation identified here has been previously reported in a family with MODY in Dresden, Germany (neonatal hypoglycaemia was not reported).5 The resulting truncated protein lacks ligand binding and transactivation domains.5,6 According to American College of Medical Genetics guidelines adapted for use in the UK,7 the mutation was deemed pathogenic by three pieces of evidence: one very strong (PVS1: a null variant in a gene where loss of function is a known mechanism of disease) and two moderate (PM2: absent from controls in the gnomAD database,8 and PS4_mod: variant previously identified in two or more unrelated affected individuals and not in the gnomAD database).
Hyperinsulinaemic hypoglycaemia may be due to structural, pharmacologic or genetic causes. Hypoglycaemia due to HNF4A is relatively mild and responsive to diazoxide.2 Our patient’s indolent clinical course and strong family history led us to suspect a genetic cause at the outset. Otherwise, suspected hyperinsulinaemic hypoglycaemia would be investigated by a prolonged fast and, if appropriate, pancreatic imaging and measurement of sulphonylurea concentrations.
Conclusion
To our knowledge this is the first case reported where increased function of HNF4A resulting in recurrent hypoglycaemia that has been sustained throughout life into the seventh decade. It is striking that the same genotype led to profoundly different manifestations in his kindred. 
References
1 Pearson ER, Boj SF, Steele AM et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 2007; 4: e118.
2 Flanagan SE, Kappor RR, Mali G et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol 2010; 162: 987–92.
3 Bacon S, Kyithar MP, Condron EM et al. Prolonged episodes of hypoglycaemia in HNF4A-MODY mutation carriers with IGT. Evidence of persistent hyperinsulinism into early adulthood. Acta Diabetol 2016; 53: 965–72.
4 Laver TW, Colclough K, Shepherd M et al. The common p. R114W HNF4A mutation causes a distinct clinical subtype of monogenic diabetes. Diabetes 2016; 65: 3212–7.
5 Lindner T, Gragnoli C, Furuta H et al. Hepatic function in a family with a nonsense mutation (R154X) in the hepatocyte nuclear factor-4alpha/MODY1 gene. J Clin Invest 1997; 100: 1400–5.
6 Eeckhoute J, Formstecher P, Laine B. Maturity-onset diabetes of the young Type 1 (MODY1)-associated mutations R154X and E276Q in hepatocyte nuclear factor 4alpha (HNF4alpha) gene impair recruitment of p300, a key transcriptional co-activator. Mol Endocrinol 2001; 15: 1200–10.
7 Ellard S, Baple EL, Callaway A et al. ACGS best practice guidelines for variant classification in rare disease 2020. ACGS Guidelines 2020.
8 Karczewski KJ, Francioli LC, MacArthur DG et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581: 434–43.